
Immunotherapy has emerged as a revolutionary approach in cancer treatment, offering hope of cure to numerous cancer patients. Ongoing research is combining this innovative approach with traditional cancer treatments, utilizing the patient’s immune system to combat cancer. Among various immunotherapies, we will now explore Adoptive T Cell Transfer therapy (ATC) following immune checkpoint inhibitors.
ATC therapy involves the transplantation of autologous or allogeneic T cells into cancer patients, showing promising results in recent years. The first successful clinical case of ATC therapy involved treating leukemia through allogeneic hematopoietic stem cell transplantation, demonstrating the effectiveness of T cells and the immune response against cancer cells.
This article will divide ATC therapy into two categories for discussion. Firstly, we will explore the transplantation method involving Tumor-Infiltrating Lymphocytes (TILs), and secondly, the transplantation of engineered T cells. After understanding both approaches, we will examine the current limitations faced by ATC therapy.
ATC with tumour-infiltrating lymphocytes
The use of ATC therapy with Tumor-Infiltrating Lymphocytes (TILs) began in the 1980s, initially applied to metastatic melanoma. Lymphocytes obtained from patient tumor biopsies, when administered intravenously along with IL-2, showed a therapeutic response rate of 34%. The therapeutic effect duration was 4 months, even some patients exhibited complete recovery. Additionally, when screening for neoantigen-specific TILs was introduced, the approach demonstrated very favorable outcomes in metastatic breast cancer. However, to observe therapeutic effects through ATC therapy, effector T cells demonstrating anticancer effects must be distributed within the tumor. This requirement poses challenges in applying the method to various cancer types, including solid tumors.
Engineered lymphocytes for ATC
Efforts to achieve mass proliferation of T cells capable of exhibiting cancer-specific activity led to the development of engineered lymphocytes with modified TCRs. Conventional T cells often exhibit reduced responsiveness to tumor antigens presented by MHC, which can be addressed by expressing synthetic Chimeric Antigen Receptors (CARs) capable of directly recognizing antigens on the cancer cell surface. T cells obtained from patients or allogeneic donors undergo genetic modifications for CAR expression, followed by mass proliferation. The expanded T cells are then reinfused into the patient for therapeutic purposes.
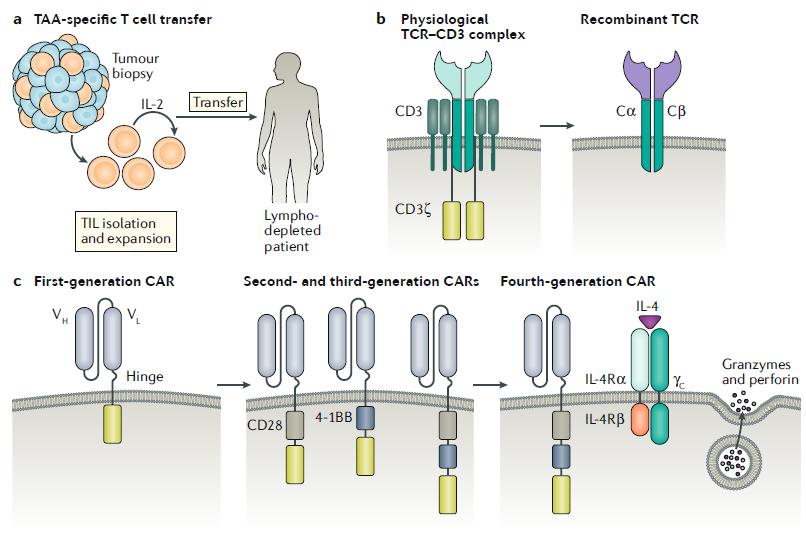
CARs are composed of an antigen-binding domain from antibodies, linked to signaling domains of the TCR and various co-stimulatory molecules. Due to the nature of cell surface signaling proteins, engineering allows the combination of various extracellular and intracellular domains, enabling the synthesis and expression of CARs tailored to specific cancers.
First-generation CAR-T cell therapies relied solely on the CD3 ζ-chain to generate TCR signals, but this design proved less effective in clinical settings due to limited T cell proliferation and reduced cytokine production. Second-generation CAR-T cells, designed to overcome these limitations, incorporate domains of CD28 and CD40 ligand along with positive regulators inducing T cell activation, resulting in improved efficacy compared to the first generation.
A notable characteristic of CAR-T cell therapy is that the target antigen presented on the cancer surface must be tissue-specific. For example, CAR-T cells targeting CD19 (predominantly expressed in B cells) are applied to B-cell lymphomas. In clinical trials, CD19-specific second-generation CAR-T cells have shown significant results in chronic lymphocytic leukemia, B-cell acute lymphoblastic leukemia, and diffuse large B-cell lymphoma, leading to FDA approval. The clinical success of these CAR-T cells was attributed to the high expression of CD19 in specific B-cell lymphomas exhibiting B-cell-specific expression. However, research is ongoing for cancer types that do not express CD19.
CAR-T therapy has demonstrated limited efficacy in solid tumors, prompting continuous efforts to overcome this limitation. Recently identified pan-cancer target B7-H3 (also known as CD276) has shown promising results in various pediatric solid tumor models. Besides direct cytolytic action, CAR-T cells can alter the tumor microenvironment and revive exhausted T cells. Preclinical models with Mucin16(MUC16ecto) specific CAR-T cells have shown promising results in ovarian cancer, with a phase I clinical trial underway for patients with ovarian cancer.
Efforts to enhance the efficacy of CAR-T cells in solid tumors also include co-expression of a chimeric cytokine receptor (4αβ), stimulating proliferation in response to IL-4 abundant in the tumor microenvironment. Preliminary studies have shown the effectiveness of this approach for CAR-T cells directed against different tumor-associated antigens (TAAs). Although other novel engineering approaches including overexpression of the transcription factor JUN are in trial, further research should be continued to expand the CAR-T therapy application scope.
Limitations and adverse effects of ATCs
The toxicity of CAR-T cell therapy varies in terms of scope and severity. The most common side effects include cytokine release syndrome (CRS) and neurotoxicity. CRS occurs due to the potent activation and proliferation of CAR-T cells and typically manifests rapidly after cell infusion. Symptoms are usually mild and flu-like, but in severe cases, it can lead to hypotension, high fever, vascular leakage, coagulation disorders, and multi-organ dysfunction. Neurological symptoms may result in encephalopathy syndrome, leading to complications such as delirium, seizures, or cerebral edema. Glucocorticoids are the primary treatment for alleviating these side effects, and the anti-IL-6 antibody Tocilizumab is used as a secondary treatment for CRS.
Specifically for CD19-specific CAR-T cell therapy, side effects may include lymphopenia and hypogammaglobulinemia, which are treated with intravenous antibody therapy similar to the treatment received by patients with underlying B-cell immune deficiencies. The mechanisms underlying these side effects are not yet clear, and additional research is underway.
ATC therapy is designed based on individualized patient treatments, leading to significant costs. The cost of CAR-T cell therapy is tens of times higher than that of immune checkpoint inhibitors, posing a significant economic burden on patients. Additionally, limited certificated facilities for CAR-T cell production make mass supply challenging, and the availability of specialized medical centers for this treatment is limited, reducing patient accessibility. Finally, the variability in the CAR-T cell manufacturing process and the absence of standardized production processes pose challenges in ensuring consistent treatment outcomes.
ATC therapy, utilizing T cells that recognize specific antigens on tumor cells, has the advantage of selectively eliminating tumor cells for cancer treatment. However, challenges such as difficulty in application to tumor types lacking specific antigens and solid tumors, along with low treatment accessibility, production volume, and process instability, limit its broader application. Overcoming these limitations could position ATC therapy as an optimized anticancer treatment tailored to individual patients, offering a potentially superior alternative to traditional anticancer therapies.
Reference : Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020 Nov;20(11):651-668.